
Department of Chemistry
 |
 |
 |
 |
 |
Tiff Walsh's research interests
van der Waals clusters
relaxation and reactivity of metal clusters
organic-inorganic interfaces
exact exchange density functional theory
polarisable potentials for molecules
van der Waals clusters
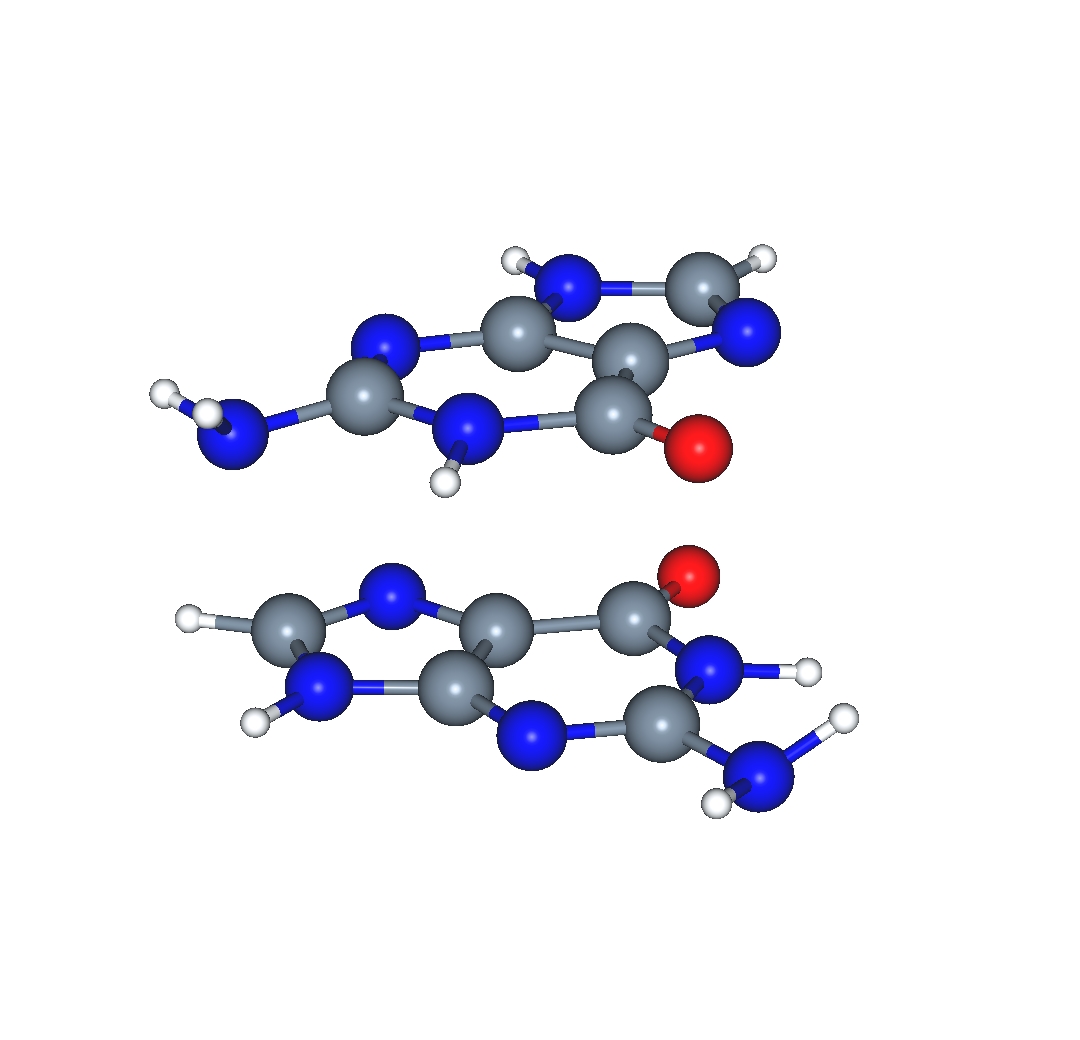
I have been investigating the use of the Wilson-Levy correlation functional as a perturbative correction to Hartree-Fock total energies. This method has so far been proven to work very well with dimers such as the guanine dimer, as is found in DNA base-pair stacking interactions.
Relaxation and reactivity of metal clusters
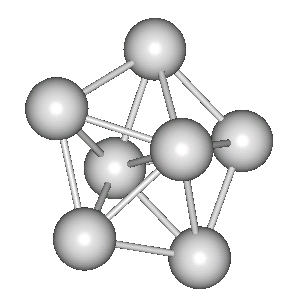
We have been using the Finnis-Sinclair potential and DFT calculations to investigate the potential energy surfaces of niobium clusters, in the size regime from N=7 -> 13 and also N=19. We have used basin-hopping to explore the potential energy surface, and have also focussed on locating the transition-states corresponding to cluster relaxation. We have used eigenvector following to calculate relaxation pathways for these relaxation mechanisms.
Click here and follow the link to view preliminary results of this work.
Organic-inorganic interfaces
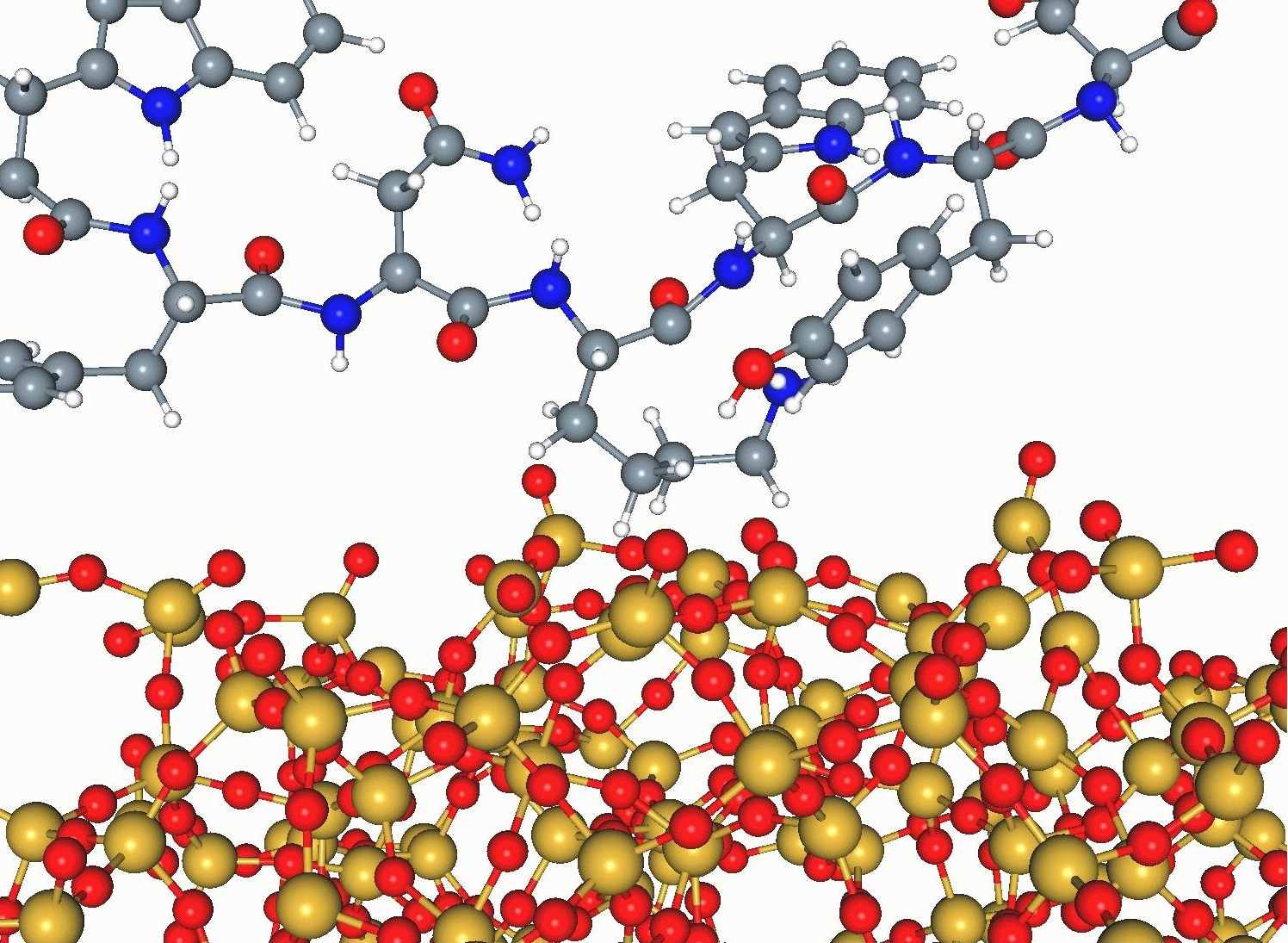
We will be developing forcefields that can describe interactions between inorganic surfaces, such as titania, silica or hydroxyapatite, and biological molecules, such as amino acids, peptides and ultimately, proteins. Our forcefields will be derived from electrostatics from ab initio calculations, and short range repulsion and dispersion interactions from plane-wave DFT calculations.
Exact exchange density functional theory
All exchange functionals in mainstream density-functional theory (DFT) do not describe exchange very well in the low-density/high-density-gradient regime. Correct description of exchange under this set of conditions is vital to describe weak interactions at intermediate separations for weakly-bound systems (where there is small but non-negligible overlap between fragments). Implementation of exact-exchange in Kohn-Sham DFT is being currently pursued.
coarse-grained potentials for polymers
van der Waals clusters
I have been investigating the use of the Wilson-Levy correlation functional as a perturbative correction to Hartree-Fock total energies. This method has so far been proven to work very well with dimers such as the guanine dimer, as is found in DNA base-pair stacking interactions.
Relaxation and reactivity of metal clusters
We have been using the Finnis-Sinclair potential and DFT calculations to investigate the potential energy surfaces of niobium clusters, in the size regime from N=7 -> 13 and also N=19. We have used basin-hopping to explore the potential energy surface, and have also focussed on locating the transition-states corresponding to cluster relaxation. We have used eigenvector following to calculate relaxation pathways for these relaxation mechanisms.
Click here and follow the link to view preliminary results of this work.
Organic-inorganic interfaces
We will be developing forcefields that can describe interactions between inorganic surfaces, such as titania, silica or hydroxyapatite, and biological molecules, such as amino acids, peptides and ultimately, proteins. Our forcefields will be derived from electrostatics from ab initio calculations, and short range repulsion and dispersion interactions from plane-wave DFT calculations.
Exact exchange density functional theory
All exchange functionals in mainstream density-functional theory (DFT) do not describe exchange very well in the low-density/high-density-gradient regime. Correct description of exchange under this set of conditions is vital to describe weak interactions at intermediate separations for weakly-bound systems (where there is small but non-negligible overlap between fragments). Implementation of exact-exchange in Kohn-Sham DFT is being currently pursued.